Cleaning validation in patch manufacturing
Mauro Scasso – Quality Assurance Supervisor
Cleaning validation is a procedure of establishing evidence that cleaning processes for manufacturing equipment prevents product contamination
This process allows us to assure that a cleaning procedure removes residues of the active pharmaceutical ingredients of the transdermal product as well as other residues or contaminants from the equipment used for the manufacturing process.
Residues shall be removed below a predetermined level in order to assure the quality of the next product on the manufacturing plan. Transdermal products can be contaminated by substances such as active pharmaceutical ingredients (API) and contaminants associated with microbes, airborne materials such as dust and decomposition residues from API especially when using campaign manufacturing.
The objective of cleaning validation is to prove that manufacturing equipment is consistently cleaned of substances and/or contaminants residues to an acceptable level, to prevent possible contamination and also cross-contamination. Cleaning validation is the documented evidence that an approved cleaning procedure will provide a high degree of certainty suitable equipment for patch manufacturing.
REGULATORY REQUIREMENTS
The local regulations at Argentina, ANMAT 3827/2018 “Guía de Buenas Prácticas de Fabricación para Elaboradores, Importadores/Exportadores de Medicamentos de Uso Humano” establishes the regulations and policies relating to pharmaceutical grade products distributed commercially within the country and it provinces:
“Cleaning validation shall be executed with the aim of ensuring the effectiveness of any cleaning procedure used for the manufacturing equipment in contact with the product”.
“Selection of residual levels shall be reasonably justified according materials used”
“Levels shall be achievable and verifiable”
“Periods of time between use and cleaning, as well as between cleaning and use shall be validated”
CLEANING VALIDATION PROGRAM
Equipment cleaning validation may be performed concurrently with actual production steps during process development and manufacturing. The validation program should be continued through full-scale commercial production. A validation program should encompass at least three consecutive successful replicates to establish that cleaning procedures are reproducibly effective. If the equipment of the similar size, design and construction is cleaned by the same procedure, studies need not be conducted on each unit as long as a total of three successful replicates are done on similar pieces of equipment; this concept is known as equipment grouping.
ESTABLISHMENT OF LIMITS
The rationale for establishing limits for product residues should be supported and based on the materials involved and their therapeutic dose. The limits should be practical, achievable, and verifiable. The approach for setting limits can be:
- Specific cleaning validation for each product
- Grouping into product families and choosing a “worst-case” product
- Grouping products according to risk, e.g. very soluble products, products with similar potency, highly toxic or difficult to detect products.
ACCEPTANCE CRITERIA
Step # | Testing Parameter | Acceptance criteria |
1 | Physical determination | The equipment should be visually clean. i.e. no residue should be visible on equipment after cleaning. |
2 | Chemical determination | Whichever is lower: a) NMT 0.1% of the normal therapeutic dose of any product to appear in the maximum daily dose of the subsequent product. b) NMT 10 ppm of any product to appear in the next product |
3 | Microbial contamination | Bacteria <50 UFC/25 cm² Yeasts and molds <5UFC/25 cm² |
CLEANING VALIDATION SCHEME
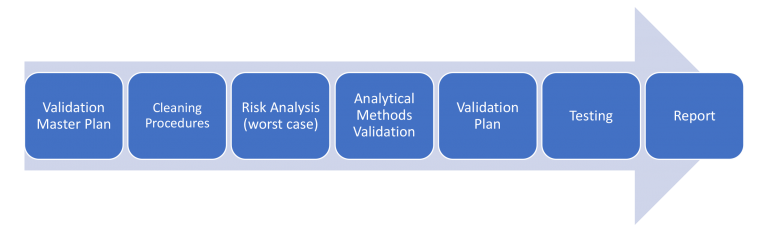
Once the Validation Master Plan and the Cleaning Procedures are getting done the next step is to evaluate the associated risk from the active pharmaceutical ingredients (APIs) from all products which pass through the same equipment line.
An approach for evaluating the Worst Case could be to evaluate solubility, toxicity and difficulty of cleaning in a risk matrix and select the worst-case API as the one that has at least 2 of 3 variables as worst case. Example:
Product | API | Solubility | Toxicity (mg / Kg) | Cleaning | S | T | C | WORST CASE |
Product 1 | API 1 | Highly soluble | v.o: 3000 (rat) | Intermediate | ||||
API 2 | Soluble | v.o: 640 (rat) | ||||||
Product 2 | API 1 | Soluble | v.o: 760 (mouse) | Hard | X | |||
API 2 | Soluble | v.o: 640 (rat) | X | |||||
Product 3 | API 1 | Soluble | v.o: 205 (rat) | Hard | ||||
API 2 | Insoluble | v.o: 395 (mouse) | X | X | X |
Analytical methods shall be validated including sampling type, swabs extraction and surfaces to evaluate. Analytical equipment widely used are HPLC / GC.
Microbiologic methods shall be compendial and sampling requires a high grade of training by the operator.
The Validation Plan must include:
- Description of the equipment on which the cleaning is to be executed
- Materials to be used
- Limits or acceptance criteria
- Parameters to be controlled or monitored
- Analytical method to be used
- Replicates to be performed
- Sampling type
- Number of samples, labeling, etc.
It is good practice to include diagrams or photos indicating the points of defined sampling. Templates or forms to be completed during execution must also be included (if not included in a SOP).
The most common sampling methods used for Testing are swabbing and rinsing since both can detect soluble and insoluble residues. Any sampling method used must be capable of quantitatively measuring the amount of residue remaining on the surface of equipment after a cleaning operation.
Subscribe to
#AmarinNews
Partner with us!
We offer expertise and experience, together with flexibility and the ability to adapt to your needs.
- info@amarintech.com.ar
- +54 11 4588-6500
- Sanchez 2045 (C1416BQG), Buenos Aires, Argentina.
