Validación de limpieza en la fabricación de parches transdermales
Mauro Scasso – Supervisor Aseguramiento de Calidad
La validación de limpieza es el procedimiento que establece evidencia respecto de los procesos de limpieza sobre los equipos de fabricación en pos de prevenir la contaminación del producto.
Este procedimiento nos permite asegurar que el proceso de limpieza elimine los residuos de los principios activos farmacéuticos del producto transdérmico, así como otros residuos o contaminantes de los equipos utilizados para el proceso de fabricación.
Los residuos se eliminarán por debajo de un nivel predeterminado para asegurar la calidad del siguiente producto en el plan de fabricación. Los productos transdérmicos pueden estar contaminados por sustancias tales como ingredientes farmacéuticos activos (IFA) y contaminantes asociados con microbios, materiales transportados por el aire como polvo y residuos de descomposición del IFA especialmente cuando se utiliza la fabricación de campañas.
El objetivo de la validación de la limpieza es demostrar que los equipos de fabricación se limpian constantemente de sustancias y/o residuos de contaminantes a un nivel aceptable, para evitar una posible contaminación y también la contaminación cruzada. La validación de la limpieza es la evidencia documentada de que un procedimiento de limpieza aprobado proporcionará con un alto grado de certeza un equipo adecuado para la fabricación de parches.
Requisitos reglamentarios
La normativa local en Argentina, ANMAT 3827/2018 “Guía de Buenas Prácticas de Fabricación para Elaboradores, Importadores / Exportadores de Medicamentos de Uso Humano” establece las normas y políticas relativas a los productos de grado farmacéutico distribuidos comercialmente en el país y sus provincias:
“La validación de la limpieza se realizará con el objetivo de asegurar la efectividad de cualquier procedimiento de limpieza utilizado para los equipos de fabricación en contacto con el producto”.
"La selección de niveles residuales se justificará razonablemente según los materiales utilizados"
"Los niveles deberán ser alcanzables y verificables"
“Se validarán los periodos de tiempo entre uso y limpieza, así como entre limpieza y uso”
Programa de validación de limpieza
La validación de la limpieza del equipo se puede realizar al mismo tiempo que los pasos de producción reales durante el desarrollo del proceso y la fabricación. El programa de validación debe continuar a través de la producción comercial a gran escala. Un programa de validación debe abarcar al menos tres réplicas exitosas consecutivas para establecer que los procedimientos de limpieza son efectivos en su repetición. Si el equipo de tamaño, diseño y construcción similares se limpia con el mismo procedimiento, no es necesario realizar estudios en cada unidad siempre que se realicen un total de tres réplicas exitosas en equipos similares; este concepto se conoce como agrupación de equipos.
Establecimiento de límites
La justificación para establecer límites para los residuos de productos debe sustentarse y basarse en los materiales involucrados y su dosis terapéutica. Los límites deben ser prácticos, alcanzables y verificables. El enfoque para establecer límites puede ser:
- Validación de limpieza específica para cada producto
- Agruparse en familias de productos y elegir un producto en el "peor de los casos"
- Agrupar productos según el riesgo, p. Ej. productos muy solubles, productos con potencia similar, productos altamente tóxicos o difíciles de detectar.
Criterios de aceptación
Paso # | Parametro de prueba | Criterios de aceptacion |
1 | Determinación física | El equipo debe estar limpio visualmente, es decir, no debe haber ningún residuo visible en el equipo después de la limpieza. |
2 | Determinación química | El que sea menor: a) No más de 0,1% de la dosis terapéutica normal de cualquier producto que aparezca en la dosis máxima diaria del producto posterior. b) No más de 10 ppm de cualquier producto que aparezca en el siguiente producto |
3 | Contaminación microbiana | Bacterias <50 UFC / 25 cm² Levaduras y mohos <5UFC / 25 cm² |
Esquema de validación de limpieza
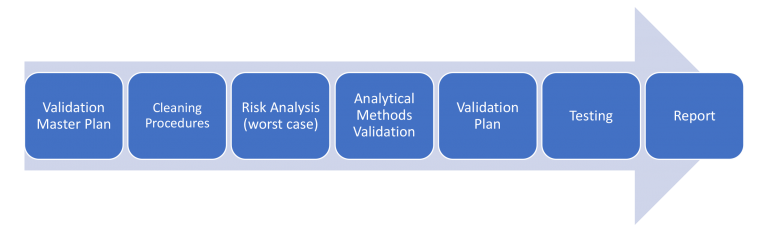
Una vez que se completan el Plan Maestro de Validación y los Procedimientos de Limpieza , el siguiente paso es evaluar el riesgo asociado de los ingredientes farmacéuticos activos (IFA) de todos los productos que pasan por la misma línea de equipos.
Un enfoque para evaluar el peor de los casos podría ser evaluar la solubilidad, la toxicidad y la dificultad de limpieza en una matriz de riesgo y seleccionar el IFA del peor de los casos como aquel que tiene al menos 2 de 3 variables como el peor de los casos. Ejemplo:
Producto | API | Solubilidad | Toxicidad (mg / Kg) | Limpieza | S | T | C | Peor caso |
Producto 1 | API 1 | Altamente soluble | v.o: 3000 (rata) | Intermedia | ||||
API 2 | Soluble | v.o: 640 (rata) | ||||||
Producto 2 | API 1 | Soluble | v.o: 760 (ratón) | Difícil | X | |||
API 2 | Soluble | v.o: 640 (rata) | X | |||||
Producto 3 | API 1 | Soluble | v.o: 205 (rata) | Difícil | ||||
API 2 | Insoluble | v.o: 395 (ratón) | X | X | X |
Los métodos analíticos deben validarse , incluido el tipo de muestreo, la extracción de hisopos y las superficies a evaluar. Los equipos analíticos ampliamente utilizados son HPLC / GC.
Los métodos microbiológicos deben ser un compendio y el muestreo requiere un alto grado de capacitación por parte del operador.
La El Plan de Validación debe incluir:
- Descripción del equipo sobre el que se va a realizar la limpieza
- Materiales a utilizar
- Límites o criterios de aceptación
- Parámetros a controlar
- Método analítico a utilizar
- Replicaciones a realizar
- Tipo de muestreo
- Número de muestras, etiquetado, etc.
Es una buena práctica incluir diagramas o fotografías que indiquen los puntos de muestreo definidos. También se deben incluir las plantillas o formularios que se completarán durante la ejecución (si no se incluyen en un POE).
Los métodos de muestreo más comunes utilizados para las pruebas son el hisopado y el enjuague, ya que ambos pueden detectar residuos tanto solubles como insolubles. Cualquier método de muestreo utilizado debe ser capaz de medir cuantitativamente la cantidad de residuos que quedan en la superficie del equipo después de una operación de limpieza.
Suscribite a
#AmarinNews
Error: Formulario de contacto no encontrado.
¡Asóciese con nosotros!
Ofrecemos experiencia y “know how”; y al mismo tiempo, flexibilidad y adaptabilidad a los requerimientos de nuestros clientes.
- info@amarintech.com.ar
- +54 11 4588-6500
- Sanchez 2045 (C1416BQG), Buenos Aires, Argentina.
