Los tiempos de la industria
Mailén Agüero – Analista de Desarrollo de Negocios
Francisco Stefano – Director
La llegada de un medicamento al mercado cuenta con muchos desafíos.
El desarrollo de un medicamento es un proceso largo y complejo, que puede durar entre 10 y 15 años desde el descubrimiento de una molécula candidata a la autorización de comercialización por parte de la agencia reguladora.
Sin embargo, cuando estamos hablando de un producto farmacéutico genérico se tarda entre 6 y 7 años aproximadamente en salir al mercado.
Todo medicamento debe atravesar un riguroso proceso de investigación y calidad. Este es un requisito fundamental para su posterior comercialización y administración en personas.
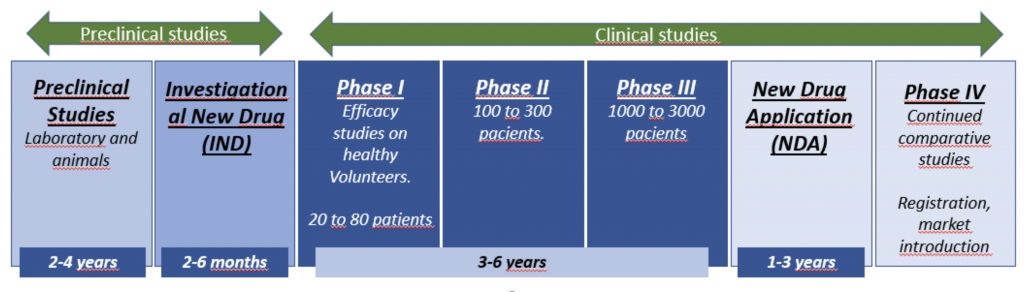
- Pruebas preclínicas
- Solicitud de nuevo fármaco en investigación (IND)
- Ensayos clinicos: Fase I, II y III
- Solicitud de nuevo fármaco (NDA)
- Estudios de fase IV
Pruebas preclínicas
Una compañía farmacéutica realiza ciertos estudios antes de que el futuro fármaco se administre a un ser humano. Se deben realizar estudios de laboratorio y en animales para demostrar la actividad biológica del fármaco contra la enfermedad objetivo. También se debe evaluar la seguridad del fármaco. Estas pruebas toman en promedio 3 1/2 años.
Solicitud de nuevo fármaco en investigación (IND)
La compañía farmacéutica presenta una solicitud (Investigational New Drug, IND) ante la agencia regulatoria para comenzar a probar el fármaco en personas. La IND entra en vigencia si la agencia regulatoria no la desaprueba dentro de los 30 días. El IND debe incluir la siguiente información: los resultados de experimentos anteriores; cómo, dónde y por quién se llevarán a cabo los nuevos estudios; la estructura química del compuesto; cómo se cree que funciona en el cuerpo; cualquier efecto tóxico encontrado en los estudios con animales; y cómo se fabrica el compuesto.
Ensayos Clínicos
Fase 1: Fase I: los estudios de fase I suelen ser las primeras pruebas de un fármaco en desarrollo en voluntarios sanos. Estos estudios involucran alrededor de 20 a 80 voluntarios. Las pruebas determinan la seguridad de un fármaco, incluido el rango de dosis seguro, además de cómo se absorbe, distribuye, metaboliza y excreta el fármaco, y la duración de su acción. Los ensayos de fase I toman en promedio 1 año.
Fase 2: estos estudios se realizan en pacientes con la enfermedad para la que está destinado el medicamento. Esta fase suele estar diseñada para identificar cuáles son las dosis mínimas y máximas. Los ensayos generalmente involucran de 100 a 300 pacientes voluntarios y tienen un diseño controlado. Se realizan para evaluar la eficacia del fármaco. La fase II suele durar unos 2 años.
Fase 3: estos son los grandes ensayos aleatorios definitivos que se envían a la agencia regulatoria para obtener la aprobación de un medicamento. Esta fase examina la eficacia y la seguridad (eventos adversos) del nuevo fármaco. Los ensayos de fase III generalmente involucran de 1000 a 3000 pacientes en clínicas y hospitales. A los pacientes se les suele pedir una lista de posibles efectos secundarios, a menudo derivados de lo observado en los estudios de fase II. Los pacientes también son libres de informar cualquier otro efecto secundario que ocurra mientras toman el nuevo fármaco o el placebo (la "píldora de azúcar" que se administra a un porcentaje de pacientes en un estudio de prueba). La Fase III toma en promedio 3 años.
Solicitud de nuevo fármaco (NDA)
Después de los ensayos clínicos de fase III, se analiza todos los datos de los estudios y presenta una NDA ante la agencia regulatoria (siempre que los datos parezcan demostrar la seguridad y eficacia del fármaco). La NDA contiene todos los datos recopilados hasta la fecha sobre el medicamento. (Un NDA generalmente consta de al menos 100,000 páginas). El tiempo promedio de revisión de NDA es de 30 meses (2 1/2 años).
Estudios de fase IV
La fase IV es cualquier recopilación organizada de datos de pacientes que están tomando un medicamento que ya recibió la aprobación de la agencia regulatoria. En los estudios de fase IV, los pacientes pueden marcar casillas en una lista (como en los estudios de fase III) o simplemente pueden informar otros síntomas.
Medicamentos Genericos
Debido a que los medicamentos genéricos son comparables a los medicamentos que ya están en el mercado, no se realizan ensayos clínicos para demostrar que su producto es seguro y eficaz. En su lugar, realizan estudios de bioequivalencia y presentan una solicitud abreviada de nuevo fármaco (Abbreviated New Drug Application, ANDA)
Existe una tercera opción llamada 505(b)(2) / Híbrido, para más información lo invitamo a leer el artículo de nuestro director:
“Sistemas de Administración Transdermal de Drogas y Vías Regulatorias 505(b)(2) / Híbrido.”
Suscribite a
#AmarinNews
¡Asóciese con nosotros!
Ofrecemos experiencia y “know how”; y al mismo tiempo, flexibilidad y adaptabilidad a los requerimientos de nuestros clientes.
- info@amarintech.com.ar
- +54 11 4588-6500
- Sanchez 2045 (C1416BQG), Buenos Aires, Argentina.
