Introduction:
In the pharmaceutical industry, the validation of computerized systems is essential to ensure that systems impacting the GxP (Good x Practice) environment meet applicable quality standards and regulations. This practice not only supports data integrity but also ensures patient safety and product efficacy, aspects that are the direct responsibility of each manufacturer.
What is a GxP Computerized System?
A Computerized System is defined as a system that includes hardware, software, network components, controlled functions, involved personnel, and associated documentation.
A GxP Computerized System is a system subject to GxP regulations (good manufacturing, distribution, and other applicable practices in the pharmaceutical and life sciences industry). In other words, it is a computerized system somehow involved in any of these activities. These systems must meet all applicable regulatory requirements and ensure their suitability for intended use. They encompass a wide range of systems, such as quality management systems, laboratory systems, manufacturing systems, clinical trial data management systems, manufacturing resource planning systems, etc.
Computerized System Inventory
All computerized systems that require validation must be listed, meaning they must be included in an inventory.
A system inventory is a documented record that regulated companies must maintain, which shows which systems are regulated by GxP. This inventory provides summarized information such as validation status, ownership, impact, current version, and supplier for each system. The goal is to have adequate control over the systems supporting business processes, avoiding duplication and ensuring that the information is at an appropriate level—at the system level and not individual hardware elements.
A documented inventory is fundamental for effective system management, regulatory compliance, and planning for maintenance and review.
To prepare the computerized system inventory, it is essential to follow an orderly and structured process that allows recording and managing GxP-impact systems and their associated risk levels. The following steps should be taken:
1) IDENTIFICATION OF COMPUTERIZED SYSTEMS
Conduct a thorough mapping of the company’s business processes and areas to identify all computerized systems in use.
2) GxP IMPACT ASSESSMENT (STEP A)
The GxP impact assessment involves an initial risk evaluation to determine the overall impact a computerized system may have on patient safety, product quality, and data integrity.
The following questions may serve as a guide:
N° | Questions |
1 | Does the Computerized System (CS) directly or indirectly control or monitor product quality? |
2 | Does the CS affect product registration, e.g., Drug Master File, Dossier, etc.? |
3 | Does the CS generate, manage, calculate, or store GxP-relevant data covered by regulations such as 21 CFR P11, Annex 11 EU-GMP, or others? Does it use electronic signatures? |
4 | Does the CS indicate materials to be used (e.g., raw materials, packaging materials, formulated products, clinical trial materials, etc.)? |
5 | Is it used for RECALL, stock traceability, or product history? |
6 | Does the CS maintain information on stock, product status, location, or shelf life? |
7 | Are data from the CS used to support product release? |
8 | Is it related to reconciliation, partial component usage, or split batches? |
9 | Is it related to labeling, coding materials, final products, or packaging components (e.g., identification labels)? |
10 | Does the CS affect or impact product quality (purity, potency, sterility, efficacy)? |
11 | Is the CS used to manage qualification data of personnel working in production, warehouse (GMP-related), or quality control? |
12 | Does the CS supervise or control the distribution, storage, or transport of products or their environmental conditions? |
13 | Does the CS influence the maintenance and calibration of equipment (managing orders, certificates, test planning, limits, etc.)? |
14 | Does the CS generate, manage, or store documents such as SOPs? |
If at least one answer to the Step A questions is YES, the system has GxP impact. Then, the level of impact is determined.
Determining the Level of GxP Impact
This evaluation seeks to determine the level of impact of a GxP System, considering process complexity and system complexity, novelty, and use. The GxP impact level determines the formality and level of documentation required for subsequent stages.
To determine the GxP Impact Level, the following categorization can be used:
- High: If the system has a direct impact on product quality, patient safety, or data integrity.
Generally, high-impact systems include those that:- Generate, manipulate, or control data supporting regulatory safety and efficacy submissions.
- Control critical parameters or data used at any stage, including preclinical, clinical, development, and manufacturing.
- Control or provide data for product release.
- Control data required for product recalls.
- Control records or notifications of adverse events or complaints.
- Support pharmacovigilance.
- Medium: If the system has a direct impact on regulations but not on product quality, patient safety, or data integrity.
- Low: If the system has an indirect impact on product quality, patient safety, or data integrity.
3) SOFTWARE COMPLEXITY EVALUATION – GAMP CATEGORIZATION (STEP B)
Software complexity evaluation, in the context of GAMP categorization, involves classifying software and hardware components based on risk, which increases as one moves from standard to customized software/hardware.
Software products are generally classified into categories, from 1 to 5:
Category 1: Infrastructure Software
These are elements that collectively form an integrated environment, enabling the execution and support of various IT services and systems.
These components include:
- Operating Systems (MS Windows, MacOS, Linux, Unix, etc.).
- Database Engines (SQL, Oracle, PostgreSQL, etc.).
- Programming Languages.
- Spreadsheets and desktop applications (Excel, Word, Outlook, etc.).
- Antivirus.
Category 3: Non-Configured Systems
- Includes “Off-the-Shelf” commercial systems:
- Non-configurable software.
- Configurable software without specific parameterization (used in a standard or default way).
- Statistical calculation software.
- Non-parameterizable data acquisition systems.
Category 4: Configured Systems
Systems that have been configured to meet the specific needs of the process or to interface with other IT systems.
Category 4 systems include:
- Standard systems with configurable functionalities to meet the business processes of the user area.
- Configurable “off-the-shelf” systems.
- Widely recognized world-class systems in the market (LIMS, MES, ERP, etc.).
- A configurable system categorized as Category 3 for lacking specific parameterization that is configured at any point in its lifecycle must be reclassified as Category 4.
Category 5: Customized Systems
Includes customized systems, as well as those developed specifically for the process they will serve.
Category 5 includes:
- Standard systems with configurable functionalities to meet the business processes of the user area, along with custom developments.
- Internal system developments.
- External developments or software not widely recognized in the industry (or from other industries).
- In summary, the evaluation of software complexity within the GAMP framework involves classifying the software into one of these categories based on its use and characteristics.

4) GENERAL EVALUATION OF SYSTEM IMPACT
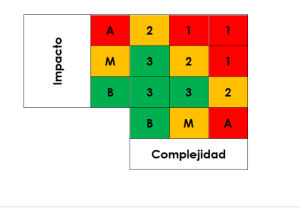
To perform a total system impact evaluation, the GxP impact evaluation from Step A and the software complexity evaluation from Step B are considered. The total risk level of the system is calculated according to the defined risk matrix:
- Class 1: High Risk
- Class 2: Medium Risk
- Class 3: Low Risk
5) DOCUMENTATION AND INVENTORY MANAGEMENT
Finally, we must integrate the information collected into a formal record, using electronic tools that allow for efficient management and periodic updates.
Define a standardized format for the inventory, including sections for each of the elements mentioned above.
Establish a schedule for reviewing and updating the system inventory, ensuring that any changes to the systems or their attributes are promptly reflected.
6) COMMUNICATION AND TRAINING
It is important to ensure that the personnel involved in system management are informed about the importance of the inventory and related processes.
Conduct training to ensure that all responsible parties understand how to effectively document and maintain the inventory.
CONCLUSION
With the information obtained through these steps, a documented and comprehensive inventory of computerized systems requiring validation is created. This inventory is key to ensuring effective control and compliance with GxP regulations, contributing to transparency, validation status control, and strategic planning for system management, facilitating audits and internal evaluations.
Implementing or reviewing the computerized system inventory is a fundamental practice to ensure quality in the pharmaceutical industry. While the validation process can be rigorous, proper preparation, management, and maintenance of this document are essential.
